二、材料与方法
(一)实验数据与软件
我们从 GSE 中筛选物种为 Homo sapiens ,类型为 Expression profiling by array,标题中同时包含乳腺或卵巢(breast|ovarian)和癌(cancer|adenocarcinoma|carcinoma|tumor)的数据集,共 1 187个(截至 2018 年 04 月 17 日)。
我们从 Broad 研究所网站(http://www.broadinstitute.org/gsea/index.jsp)下载了 GSEA 软件(Java 实现版本,javaGSEA 3.0)和基因集数据库 MSigDB (Liberzon et al. 2011) (Molecular Signatures Database)v6.1(共包含 17 786个 基因集)。
所有的数据分析均在 R 语言 3.4.4 中完成。
(二)实验方法
1. rGEO算法实现
为了确定 SOFT 文件的平台表格中每一列储存的 ID 来自哪一个数据库,我们通过正则表达式去挖掘列名和描述中的信息,比如既包含 entrez、又包含 id 的列储存的应该就是 Entrez Gene ID,同时对所有符合条件的列的列名和描述进行人工复查,剔除可能有问题的情况,比如在上述条件下额外包含 unigene 的列实际上储存的是 UniGene (Pontius, Wagner, and Schuler 2003) ID,此时规则就应该修改为,既包含 entrez、又包含 id 且不包含 unigene。
在实际操作中,我们发现有时候很难区分某一列储存的到底是 Entrez Gene ID 还是 HUGO 基因符号,注意到这两者之间没有任何重叠,我们将其合并为一个新的 ID——entrez_or_symbol,该 ID 可用于将这些难以界定的列对应到 HUGO 基因符号。同理,我们从 INSDC (Nakamura, Cochrane, and Karsch-Mizrachi 2013) accession(有时也被称为 GenBank (Benson 2004) accession)和 RefSeq (O’Leary et al. 2016) accession (nucleotide)合并得到 genbank 这一新 ID。
就这样,我们为常见数据库的 ID 都生成了相应的规则,将其应用到具体的平台表格则可以知道特定数据库的 ID 存储于哪一列。对于未来新增加的数据,可能会出现新的特例,但是我们制定的这些规则在大部分情况下还是适用的,同时这些规则具有很好的可扩展性,可以在未来进行定期增量更新。
接下来我们需要将其它数据库的 ID 对应到 HUGO 基因符号,如图 3 所示。我们从 HGNC (Yates et al. 2017)(https://www.genenames.org/help/statistics-downloads)下载了 hgnc_complete_set.txt 文件,实现了从常见数据库的 ID 到 HUGO 基因符号之间的对应,具体包括 Entrez Gene ID、Ensembl gene ID、INSDC accession、RefSeq accession(nucleotide)。我们还从 NCBI(ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2unigene)下载了 gene2unigene.tsv 文件,其中包含了 Entrez Gene ID 与 UniGene ID 之间的对应关系,结合前文,就实现了 UniGene ID 与 HUGO 基因符号之间的对应。至此,经过一到两次转换即可将 SOFT 文件中的探针对应到 HUGO 基因符号。
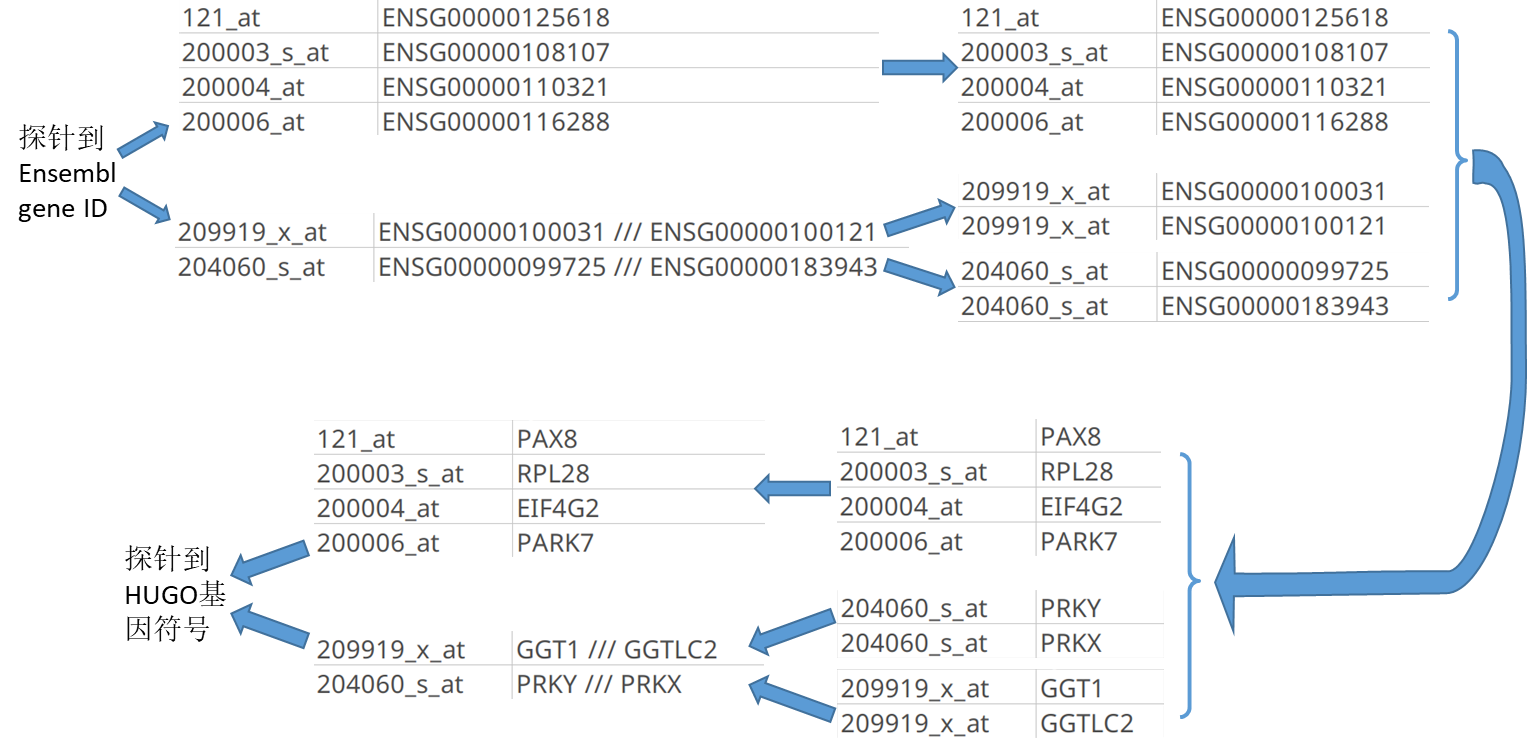
图 3: 将探针对应到 HUGO 基因符号(以通过Ensembl Gene ID 为例)
但是一个麻烦在于这些对应关系中经常包含一对多的情况。javaGSEA 允许一个探针对应到一个或多个 HUGO 基因符号,但是直接将一对多对应中的其它数据库的 ID 替换为 HUGO 基因符号会造成很大的计算开销。为此,我们首先将这些一对多对应拆分成多个一对一对应,然后把它们和剩下的一对一的对应合并(前文提到的文件中也存在一对多对应,我们就是通过这两步得到由其它数据库的 ID 到 HUGO 基因符号的一对一对应),接下来将其它数据库的 ID 对应到 HUGO 基因符号,最后将重复的一对一对应折叠成一对多对应并与剩下的一对一对应合并,如图 3 所示。
在这个过程中的另一个麻烦在于 SOFT 文件的混乱。比如,在前文提到的一对多对应中,不同平台表格中用于分割 ID 的间隔符就不统一,有 ,,///, ,等。我们巧妙地利用正则表达式中的反向匹配规则,不去拘泥于分析确切的间隔符,而是将不会在 ID 中出现的字符一律视为间隔符。例如 Entrez Gene ID 完全由数字组成,那么任何非数字的字符均会被视为间隔符。
我们将上述过程中用到的 R 语言代码分割成相对独立的函数,并在设计的过程中加入了可扩展性和重用性的考虑,这样既有助于将来的更新,也方便其它研究者完成类似的任务。
2. qGSEA软件实现
qGSEA 基于 Shiny 而开发,有两种运行模式——A 和 B,其中模式 B 又分为 B-1 和 B-2 两步。
在模式 A 中,我们根据用户提供的 GSE accession 计算出 matrix 文件和 SOFT 文件的链接,然后用 R 语言内置的函数去下载上述文件,接下来我们在后台调用 rGEO 来完成探针到 HUGO 基因符号的对应,最后生成 javaGSEA 的输入文件并将其写入用户指定的输出文件夹中(选择文件或文件夹的功能由 shinyFiles 包提供)。
在 B-1 中,我们根据用户提供的 GSE accession,使用 shinyjs 包提供的 runjs() 函数运行 JavaScript 代码,实现实时更新 Download matrix file 和 Download SOFT file 这两个按钮对应的链接。同时修改按钮的 onclick() 句柄,使其触发按钮内链接文字(<a> 元素)的 click() 事件,从而实现单击按钮或按钮内的文字均能下载文件的效果。在 B-2 中,我们仍需要用户提供 GSE accession,这样一方面便于确定生成文件的文件名,另一方面也可以验证用户是否选择了正确的原始数据文件(这里我们假定用户不会修改 B-1 中下载的原始数据的文件名,对于确实需要修改文件名的高级用户,可以直接使用 rGEO 中提供的接口),其它方面与模式 A 相似。
在运行过程中出现错误后的提示信息均使用 JavaScript 来实现动态更新。由于文件(夹)选择控件目前还未支持 Shiny 控件的全部标准,而且同时显示太多信息容易干扰用户的决策,大部分控件的提示信息都是在点击 Run 之后才会更新。但是在 B-1 中没有 Run 按钮,故我们将 GSE accession 的提示信息设置为及时更新(在输入框的状态发生变化后就会立即更新)。
3. GSEA及其结果的整合
我们使用 MSigDB 中的 13 个基因集集合(collection),如表 1 所示,以 LEM4 的表达量作为表型标签,Metric 选择 Pearson,其它参数均选用默认值。因为基于重排表型的显著性估计方法能很好地控制假阳性,而且我们这里的主要目的是生成假设,根据 GSEA 团队的推荐,我们用 FDR < 0.25 来判定统计显著性。但使用这样较大的 FDR 阈值之后,被判定为统计显著的基因集的 FDR 之间会存在较大差异,而仅使用某一基因集表现为统计显著的数据集数目 n,来评估其与 LEM4 之间的相关性则忽略了这一差异。于是,在计算 n 时,我们减去了筛选出的基因集的 FDR 的总和,使得其含义变为某一基因集表现为真阳性的数据集数目的估计值。这样可以给予 FDR 小的基因集更大的权重,从而加强统计分析的效度。
| 名称 | 描述 |
|---|---|
| h | 特征(hallmark)基因集 (Liberzon et al. 2015) |
| c1 | 位置基因集 |
| c2.cgp | 化学和遗传学扰动 |
| c2.cp.biocarta | BioCarta 基因集 |
| c2.cp.kegg | KEGG 基因集 |
| c2.cp.reactome | Reactome 基因集 |
| c3.mir | microRNA 靶基因 |
| c3.tft | 转录因子靶基因 |
| c4.cgn | 癌症基因邻居 |
| c4.cm | 癌症模块 |
| c5 | GO 基因集 |
| c6 | 癌基因标志(signature) |
| c7 | 免疫标志 |
(三)实验流程图
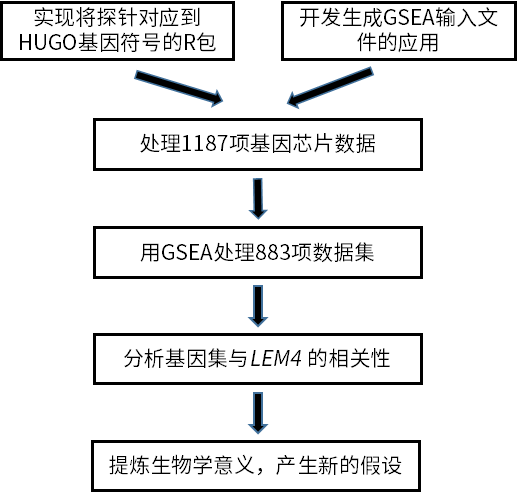
实验流程图
References
Benson, Dennis A. 2004. “GenBank.” Nucleic Acids Research 33 (Database issue): D34–D38. https://doi.org/10.1093/nar/gki063.
Liberzon, Arthur, Chet Birger, Helga Thorvaldsdóttir, Mahmoud Ghandi, Jill P. Mesirov, and Pablo Tamayo. 2015. “The Molecular Signatures Database (MSigDB) hallmark gene set collection.” Cell Systems 1 (6): 417–25. https://doi.org/10.1016/j.cels.2015.12.004.
Liberzon, Arthur, Aravind Subramanian, Reid Pinchback, Helga Thorvaldsdóttir, Pablo Tamayo, and Jill P. Mesirov. 2011. “Molecular signatures database (MSigDB) 3.0.” Bioinformatics (Oxford, England) 27 (12): 1739–40. https://doi.org/10.1093/bioinformatics/btr260.
Nakamura, Yasukazu, Guy Cochrane, and Ilene Karsch-Mizrachi. 2013. “The International Nucleotide Sequence Database Collaboration.” Nucleic Acids Research 41 (D1): D21–D24. https://doi.org/10.1093/nar/gks1084.
O’Leary, Nuala A., Mathew W. Wright, J. Rodney Brister, Stacy Ciufo, Diana Haddad, Rich McVeigh, Bhanu Rajput, et al. 2016. “Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation.” Nucleic Acids Research 44 (D1): D733–D745. https://doi.org/10.1093/nar/gkv1189.
Pontius, J U, L Wagner, and G D Schuler. 2003. “UniGene: a unified view of the transcriptome.” The NCBI Handbook, no. 21: 1–12. https://www.ncbi.nlm.nih.gov/books/NBK21083/.
Yates, Bethan, Bryony Braschi, Kristian A. Gray, Ruth L. Seal, Susan Tweedie, and Elspeth A. Bruford. 2017. “Genenames.org: the HGNC and VGNC resources in 2017.” Nucleic Acids Research 45 (D1): D619–D625. https://doi.org/10.1093/nar/gkw1033.