一、前言和文献综述
(一)GEO数据库
GEO (Barrett et al. 2012)(Gene Expression Omnibus,http://www.ncbi.nlm.nih.gov/geo/)是 NCBI (Agarwala et al. 2017)(National Center for Biotechnology Information)创建的一个国际化的公共仓库,储存着科研社区提交的高通量基因芯片和新一代测序功能基因组数据集。尽管近年来RNA-seq变得越来越流行,GEO中现存的海量基因芯片数据依然不容忽视,如图 1-A 所示,其中蕴藏着宝贵的生物学意义(后文未特殊说明时均仅讨论GEO中的基因芯片数据)。
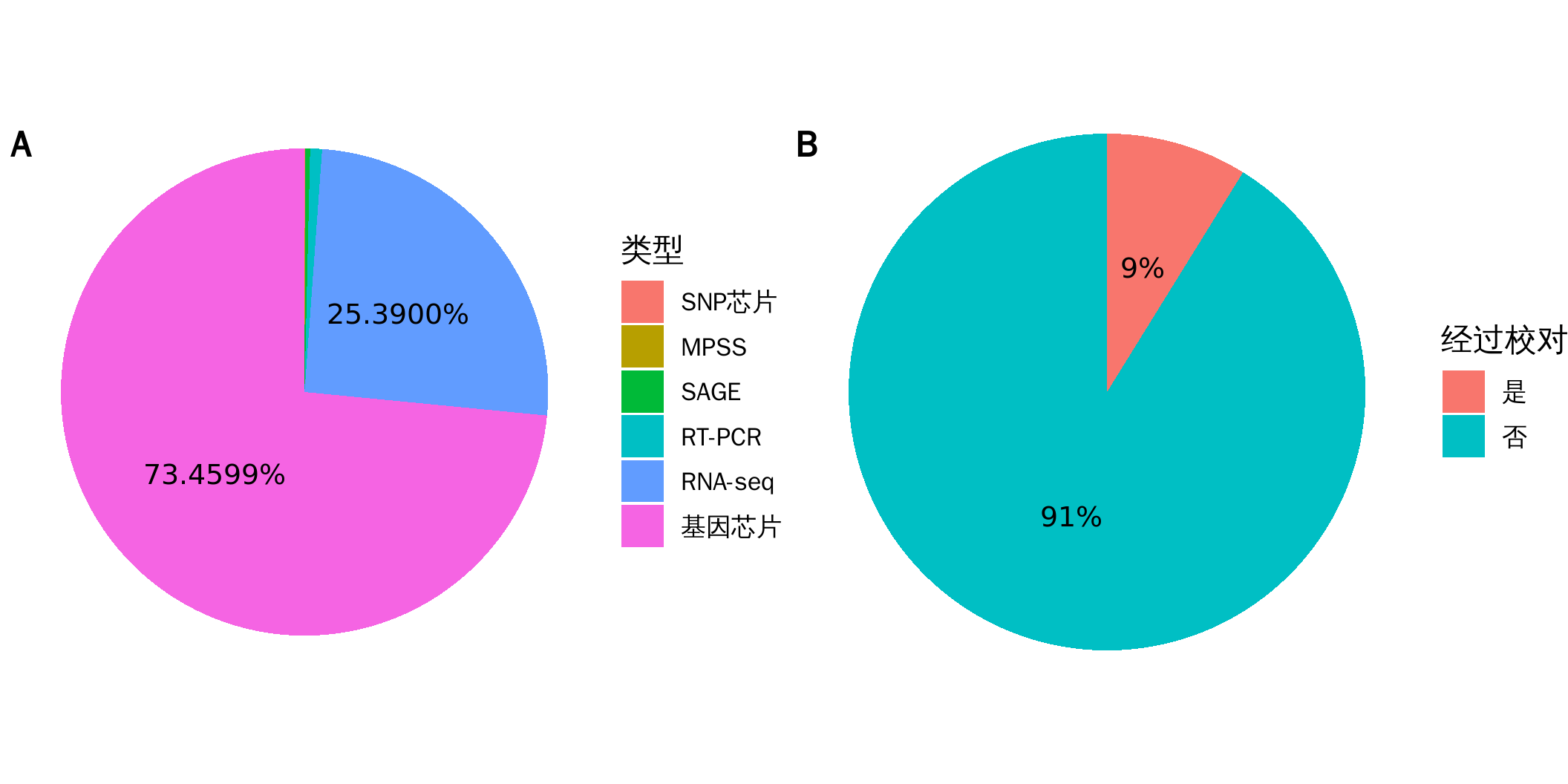
图 1: GEO 数据库中不同类型的数据分布
A. 不同类型的表达谱数据的分布(截至 2018 年 5 月)。B. GDS 在 GSE 中所占的比例(截至 2018 年 5 月 22 日)。
提交到GEO中的数据分为3种 (Edgar 2002):Platform 被赋予前缀为“GPL”的 accession,包含芯片的总结描述和定义模板的数据表格,该表格每一行代表一个特征(feature),可包含任意列注释信息(提交者提供的序列注释和追踪信息);Sample 被赋予前缀为“GSM”的 accession,包含生物学材料的描述,实验方法和可包含任意列的数据表格;Series 被赋予前缀为“GSE”的 accession(后文中称为 GSE accession),定义了一组 Sample,其被认为属于某项研究的一部分。简单来说,Platform 代表着芯片,Series 是在某一特定芯片上测量出的数据集,其中的每个个体即为 Sample。
GEO 中储存的高通量实验数据类型广泛,它们由多种方式加工、不同方法分析,这给高效的数据挖掘带来了很大的挑战 (Barrett et al. 2005) 。GEO通过一套结合了自动数据提取和人工校对的流程,从用户提交的记录中提取出 Platform 上每个 feature 的序列身份追踪信息、归一化后的表达量测量值和生物学来源及实验目的的文字描述,然后组织成一种高级数据模型——GEO DataSets(GDS,可以认为是 GSE 的一个子集),为下游的数据挖掘和展示工具奠定了基础 (Barrett et al. 2009) 。
对于每一项数据集,我们关心的信息主要有两部分:表达量数据和探针注释信息。后一部分甚至更为重要,因为只有知道了每个探针具体对应到哪个基因,我们才能诠释对表达量数据的统计学分析结果的生物学意义。GDS的数据经过了良好的注释,有着一致的格式,在“DataSet full SOFT file”中两部分信息合并成了一个表格。每项数据集的表格都包含许多注释信息,其中 Entrez Gene symbol 这一列可用于把探针对应到 HUGO 基因符号 (Yates et al. 2017) 上(Entrez Gene (Maglott et al. 2004) 以纯数字作为唯一标识符,不过每个基因都有对应的“prefered symbol”,即 HUGO 基因符号,故有时也被称为“Entrez Gene symbol”,“Entrez symbol”等)。但 GSE 中的数据格式则没有这么用户友好。表达量数据可以从“Series Matrix File”(后文称其为 matrix 文件)中方便地提取,探针注释信息储存在“SOFT formatted family file”(后文其称为 SOFT 文件)中。
SOFT (Barrett et al. 2007)(Simple Omnibus Format in Text)是 GEO 规定的一种简单、基于行、制表符分隔的文件格式,为了快速批量存放数据而设计。虽然 GEO 为 SOFT 文件中储存探针注释信息的表格(后文称其为平台表格,每一项 GSE 数据集都有对应的 Platform,其信息即主要储存于该表格中)制定了详尽的要求,但其格式依然十分混乱,为分析其中的信息带来了很大不便。GEO 只是提出了最低的要求,每一个 SOFT 文件的平台表格必须包含名为“ID”的列(储存探针名称),同时还提供了许多可选的其它列名,如果文件中出现这些列,则必须储存 GEO 规定的信息。除此之外,平台表格中可以包含任意数量的列,它们可以有着任意的列名,储存任意的信息。这就使得确定其中每一列储存的 ID(identifier,标识符)来自哪一个数据库变得非常棘手,我们可以利用的信息只有列名(name)和简短的描述(description),后文将其称为元信息(有的列储存的是 accession,后文中不引起歧义时不再作此说明)。
雪上加霜的是,从实际情况来看,连 GEO 提出的最低要求都没有被严格遵守。比如名为 ORF 的列,本应该储存 NCBI’s Entrez Genomes
division 鉴定的开放读码框,由于没有规定具体的标准,有些 SOFT 文件储存的是 ID(本段中特指 Entrez Gene ID),另一些储存的是 symbol(本段中特指Entrez Gene symbol,即 HUGO 基因符号)。但是,在 GPL6353 的平台表格中,这一列的描述赫然写着 Ensembl gene ID。在 Entrez Genome(https://www.ncbi.nlm.nih.gov/genome/?term=)搜索其中一项——ENSG00000177693,没有任何结果,而搜索其对应的 ID 或 symbol 都会有结果,另外有些基因是没有对应的“Ensembl gene ID”的(例如 ID 为 63、symbol 为 ACTBP3 的基因),更别提 Ensembl (Zerbino et al. 2018) 甚至都不属于 NCBI 的一部分。又比如GPL13915 的平台表格包含两个名为 GB_ACC 的列,根据描述,前一个储存“GenBank accession”,后一个储存“RefSeq ID”,先不提 GEO 关于这一列的具体要求,这种情况就连表格的最基本要求(列名必须唯一)都没能满足。其它情形就不一一列举了。
综上可见,将基因芯片数据中的探针对应到 HUGO 基因符号这一听起来理所当然的事情实际上有多么困难。为了接下来的数据分析,我们开发出了一个 R 包——rGEO,将不同平台表格的探针统一地对应到 HUGO 基因符号。我们发现,对于人类(Homo sapiens)的基因芯片数据而言,GDS 大概只占到 GSE 的1/10,如图 1-B 所示。这表明 rGEO 能够很大地促进研究者分析 GEO 中丰富的数据,从而产生新的生物学发现。
(二)GSEA及其发展
自从基因芯片技术出现以来,科学家一直很感兴趣鉴别差异表达基因和阐明相关的生物学过程。最常用的统计学方法——IGA(individual gene analysis)——评估单个基因在两组样本之间的显著性,并设定一个合适的截断阈值,从而得到一个差异表达基因的列表 (Nam and Kim 2008) 。随后这个列表通常会用来对基因本体论(Gene Ontology)或代谢通路数据库进行富集分析——通常是基于超几何分布或其二项分布近似的显著性分析——鉴别出过度存在(over-represented)的、行使相似生物学功能的基因类别 (Irizarry et al. 2009) 。
IGA 的主要问题在于使用了截断阈值。一方面,阈值——通常是任意选定——显著影响富集结果 (Breslin, Edén, and Krogh 2004) ,甚至会严重改变最终的生物学结论 (Pan, Lih, and Cohen 2005) 。另一方面,严格的阈值会抛弃许多表达量改变微弱、但有生物学意义的基因,从而减弱统计功效 (Ben-Shaul, Bergman, and Soreq 2005) 。而很多重要的生物学过程,例如代谢通路、转录程序、胁迫反应,分散在整个基因网络,在单基因水平表达量变化微弱。
Subramanian et al. (2005) 提出了 GSEA(Gene Set Enrichment Analysis)来分析基因表达数据,其专注于基因集,也就是一组共享相同生物学功能、染色体位置或者调控因子等的基因,这样能将已有的生物学知识列入考虑,从而增强了分析能力。GSEA 首先根据芯片上的每个基因与表型的相关系数(或其它排序指标)进行排序得到有序列表L,对于每一个基因集 S,通过遍历L计算出一个累加统计量(遇到在 S 中的基因时增加,遇到不在 S 中的基因时减少)。该累加统计量的最大值或最小值(取对应的绝对值较大者)即为富集分数(Enrichment Score,ES),然后通过重排(permutate)表型标签的方法估算 ES 的经验分布,得到显著性系数,最后矫正多重假设检验。与其它富集方法不同,GSEA 考虑到了实验中全部基因,而不仅仅是其中的一部分(任意选定表达量变化或统计显著性的阈值后进行截断而得到),而且通过重排表型标签计算显著性保留了基因-基因的相互关系,从而提供了更加准确的零模型(null model)。实践表明,相对于 IGA,GSEA 在不同数据集上的分析结果表现出更多的一致性和更好的可重复性,而且更加易于解释。
GSEA 推出后得到了广泛应用,成为了最流行的基因集分析方法之一 (Bevilacqua et al. 2012) ,同时也有很多研究者在此基础上做出改进。Dinu et al. (2007) 认为 GSEA 在鉴别与二态表型关联的生物学代谢通路时有着重要的局限性,故提出了 SAM-GS(Significance Analysis of Microarray to gene-set analyses),并表明其能识别出更多的真正有关联的基因集(其中一半以上基因与表型适度或强烈相关)和更少的无用基因集(其中没有基因与表型相关)。SetRank (Simillion et al. 2017) 通过舍弃那些仅仅因为与其它基因集重叠而被认为显著的基因集,减少了很多假阳性。PAEA (Clark et al. 2015) (Principal Angle Enrichment Analysis)使用降维和多变量方式进行基因集富集分析。GSEA-SNP (Holden et al. 2008) 将 GSEA 的思想引入到 GWAS(genome-wide SNP association studies)中,除了能鉴别作用微弱的标记物,还有助于鉴别与疾病关联的 SNP 和代谢通路,并理解其背后的生物学机制。
GSEA 的一大缺点是在估算统计显著性和矫正多重检验这一步,在重排后的数据集上产生了巨大的计算开销。为了实现有效的大尺度转录组数据分析,paraGSEA (Peng et al. 2017) 通过优化将时间复杂度从 O(mn)降到了 O(m+n)(m 是基因集长度,n 是基因表达谱的长度),实现了 100 倍的性能提升,在进一步并行加速之后,整个 LINCS(Library of Integrated Network-based Cellular Signatures)一期数据集(GSE92742)的分析时间减少到了 120 小时(96 核工作站)。PAGE (Kim and Volsky 2005) (parametric analysis of gene set enrichment)改进了 GSEA 中的统计模型,使用正态分布来进行统计分析,省去了重复计算,能检测出大量显著改变的基因集,且其 P 值比 GSEA 算出的结果要低。
同时还有许多将 GSEA 的思想拓展到RNA-seq数据分析的尝试。SeqGSEA (Wang and Cairns 2014) 使用 DESeq (Love, Huber, and Anders 2014) 分析差异表达(differential expression)、DSGSeq (Wang et al. 2013) 分析差异剪接(differential splicing),然后计算整合之后的得分(gene scores),以此进行基因集富集分析,最后用重排表型的方法计算 P 值,其缺点是每种类别至少需要 5 个样本。尽管近年来测序费用大大降低,对大部分实验室来说生成能达到该要求的样本数量还是过于昂贵 (Garber et al. 2011) ,这时只能用重排基因的方式计算 P 值,由于这样打乱了基因-基因的相互关系,很容易造成假阳性。AbsFilterGSEA (Yoon, Kim, and Nam 2016) 通过计算富集分数的平均值并使用单侧检验,只保留在这种方式和常规方式中均表现为统计显著的结果,大大降低了假阳性率,为小样本的实验提供了一种替代方案。
尽管有着诸多改进,我们发现对于湿实验工作者而言,使用 GSEA 处理数据依然较为困难。以基于 Java 的桌面应用 GSEA-P (Subramanian et al. 2007) (后来该软件被改名为 javaGSEA,后文均使用此名称)为例,虽然其有着简单易用的图形化界面和详实的帮助文档,在使用自带的测试文件时给用户带来了很好的体验;但是在分析用户自己的数据(例如从 GEO 下载的公共数据)时,在最开始的一步——准备其需要的输入文件——就能难倒很多人了。具体而言,用户需要首先理解 javaGSEA 的输入文件的格式,然后再读懂 GSE 的原始数据的格式,才能完成格式转换工作。对于湿实验研究者,这意味着在 Microsoft Excel 中操作数小时,而且在 SOFT 文件的平台表格中没有提供 HUGO 基因符号时几乎无法完成;对于生物信息学家,这意味着数至数十行代码,取决于 SOFT 文件的平台表格中是否提供了 HUGO 基因符号。
为了解决这一问题,使广大研究者能够方便地使用 GSEA 这一强大的分析方法去分析 GEO 中丰富的高通量表达数据,我们推出 qGSEA(由 rGEO 提供后台支持)。其拥有基于浏览器的用户界面,用户只需要查询到感兴趣的 GSE 数据集的 accession,即可得到完全符合 javaGSEA 要求的输入文件。qGSEA 不但能使得湿实验工作者也能轻松方便地准备 javaGSEA 所需的输入文件,而且能为经验丰富的生物信息学家节约不少时间。
值得注意的是,虽然 qGSEA 给出的结果是 javaGSEA 所需的输入文件,但这并不意味着 qGSEA 的作用仅限于此。qGSEA 最大的贡献在于将 rGEO 包装成了图形化的界面,基于 GSEA 的改进软件需要的输入信息与 javaGSEA 的大致相同,只是在格式上可能存在细微的差别。只需要对 qGSEA 的代码进行微小的修改,即可以用同样简单易用的方式为这些改进软件提供输入文件。也就是说,我们开发的工具在一定程度上能从整体水平促进基因芯片数据的分析。
(三)LEM4 的功能
LEM(LAP2-Emerin-MAN1)结构域是一段保守的、长度约为 40 氨基酸残基的螺旋-环-螺旋模序,能够与自整合障碍因子(barrier-to-autointegration factor,BAF)相互作用 (Cai et al. 2001) 。BAF 是一个长度约为 89 氨基酸残基的小蛋白,在染色体聚集、细胞周期和有丝分裂末期细胞核重组装中发挥着至关重要的作用 (Segura-Totten and Wilson 2004)。BAF 会形成二聚体,通过两个对称的 DNA 结合位点非特异性地桥接双链 DNA,同时还有一个 LEM 结构域结合位点 (Cai et al. 2007) 。LEM 结构域与其它直接结合 DNA 的双螺旋模序——螺旋-延伸-螺旋(helix-extension-helix,HeH)和 SAF-Acinus-PIAS(SAP)模序——高度相关 (Brachner and Foisner 2011) 。事实上,在包含 LEM 结构域的代表性蛋白 LAP2 中,还有一段类 LEM 结构域,能够直接结合 DNA (Cai et al. 2001) 。Brachner and Foisner (2011) 认为 LEM 结构域可能是从 HeH 和 SAP 结构域,与 BAF 共同进化而来。即使是在明显没有 BAF 和核纤层蛋白的酵母中,LEM 结构域依然保守,暗示 LEM 结构域在基因组组织和细胞核结构中有着内在的重要作用 (Berk, Tifft, and Wilson 2013) 。
目前共发现了由 7 个基因编码的 9 种 LEM 结构域蛋白,构成了 LEM 家族 (Wagner and Krohne 2007) 。依据膜定位的拓扑结构等特征,该家族被划分为 3 组,其中第 III 组的 LEM 结构域位于内部,且含有许多锚蛋白重复,LEM4(由 ANKLE2 / LEM4 基因编码)即是一个代表性的例子 (Barton, Soshnev, and Geyer 2015) 。锚蛋白重复广泛存在于自然界,专门介导蛋白质相互作用,有些与人类癌症发生直接相关 (Li, Mahajan, and Tsai 2006) 。LEM4 缺乏该家族中典型的跨膜结构域,但是在靠近 N 端的位置有一段疏水链,也许能起到膜锚定作用 (Snyers et al. 2018) 。与 LAP2、Emerin 和 MAN1 等不同,LEM4 同时定位在内核膜和内质网膜上 (Barton, Soshnev, and Geyer 2015) 。Asencio et al. (2012) 发现在有丝分裂期后期和末期,LEM4 既能抑制 VRK-1 对 BAF 的激酶活性,又能促进 PP2A 复合体对 BAF 的磷酸酶活性,通过对两者的协同调控,LEM4 控制着有丝分裂末期的核膜重建过程。
实验室前期工作发现,LEM4 能够促进乳腺癌的发展,导致三苯氧胺耐药性,并降低患者的生存率。其机理包含如下两种:一是 LEM4 通过与 CDK4 和 Rb 相互作用,增加 Rb 的稳定性和磷酸化水平,从而释放 E2F,促进细胞周期蛋白 E、CDK2 和 E2F 自身的编码基因的转录,如图 2-A 所示。一是 LEM4 通过与 Aurora-A 相互作用,增强 Aurora-A 介导的 ERα 上 Ser167残基的磷酸化,从而促进细胞周期蛋白 D1 和 MYC 编码基因的转录,如图 2-B 所示。这两种机理均能推动 G1 期到 S 期的转化。
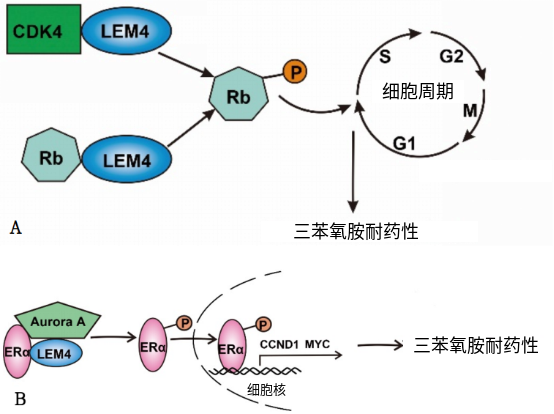
图 2: LEM4 导致三苯氧胺耐药性的分子机制
A. LEM4 调控细胞周期蛋白 D-CDK4/6-Rb 轴。B. LEM4 调控 ERα 信号通路。
为了验证实验结果和发现新的研究靶点,我们从 GEO 下载了临床病人的基因芯片数据,使用 qGSEA 转换原始数据文件,然后运用 GSEA 进行富集分析。将许多数据集的结果整合在一起后,我们构建出衡量基因集与 LEM4 表达量相关性的指数,并依此筛选出最突出的基因集。在对这些基因集的生物学意义加以分析之后,我们发现其中一部分很好地重现了前期实验或已发表论文的结果,另一些则提供了新的研究靶点。
References
Agarwala, Richa, Tanya Barrett, Jeff Beck, Dennis A. Benson, Colleen Bollin, Evan Bolton, Devon Bourexis, et al. 2017. “Database Resources of the National Center for Biotechnology Information.” Nucleic Acids Research 45 (D1): D12–D17. https://doi.org/10.1093/nar/gkw1071.
Asencio, Claudio, Iain F. Davidson, Rachel Santarella-Mellwig, Thi Bach Nga Ly-Hartig, Moritz Mall, Matthew R. Wallenfang, Iain W. Mattaj, and Mátyás Gorjánácz. 2012. “Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis.” Cell 150 (1): 122–35. https://doi.org/10.1016/j.cell.2012.04.043.
Barrett, Tanya, Tugba O Suzek, Dennis B Troup, Stephen E Wilhite, Wing-Chi Ngau, Pierre Ledoux, Dmitry Rudnev, Alex E Lash, Wataru Fujibuchi, and Ron Edgar. 2005. “NCBI GEO: mining millions of expression profiles–database and tools.” Nucleic Acids Research 33 (Database issue): D562–6. https://doi.org/10.1093/nar/gki022.
Barrett, Tanya, Dennis B Troup, Stephen E Wilhite, Pierre Ledoux, Dmitry Rudnev, Carlos Evangelista, Irene F Kim, Alexandra Soboleva, Maxim Tomashevsky, and Ron Edgar. 2007. “NCBI GEO: mining tens of millions of expression profiles–database and tools update.” Nucleic Acids Research 35 (Database issue): D760–5. https://doi.org/10.1093/nar/gkl887.
Barrett, Tanya, Dennis B. Troup, Stephen E. Wilhite, Pierre Ledoux, Dmitry Rudnev, Carlos Evangelista, Irene F. Kim, et al. 2009. “NCBI GEO: archive for high-throughput functional genomic data.” Nucleic Acids Research 37 (Database): D885–D890. https://doi.org/10.1093/nar/gkn764.
Barrett, Tanya, Stephen E. Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F. Kim, Maxim Tomashevsky, Kimberly A. Marshall, et al. 2012. “NCBI GEO: archive for functional genomics data sets—update.” Nucleic Acids Research 41 (D1): D991–D995. https://doi.org/10.1093/nar/gks1193.
Barton, Lacy J., Alexey A. Soshnev, and Pamela K. Geyer. 2015. “Networking in the nucleus: A spotlight on LEM-domain proteins.” Current Opinion in Cell Biology 34: 1–8. https://doi.org/10.1016/j.ceb.2015.03.005.
Ben-Shaul, Yoram, Hagai Bergman, and Hermona Soreq. 2005. “Identifying subtle interrelated changes in functional gene categories using continuous measures of gene expression.” Bioinformatics 21 (7): 1129–37. https://doi.org/10.1093/bioinformatics/bti149.
Berk, Jason M., Kathryn E. Tifft, and Katherine L. Wilson. 2013. “The nuclear envelope LEM-domain protein emerin.” Nucleus (Austin, Tex.) 4 (4): 298–314. https://doi.org/10.4161/nucl.25751.
Bevilacqua, Vitoantonio, Paolo Pannarale, Mirko Abbrescia, Claudia Cava, Angelo Paradiso, and Stefania Tommasi. 2012. “Comparison of data-merging methods with SVM attribute selection and classification in breast cancer gene expression.” BMC Bioinformatics 13 Suppl 7 (Suppl 7): S9. https://doi.org/10.1186/1471-2105-13-S7-S9.
Brachner, Andreas, and Roland Foisner. 2011. “Evolvement of LEM proteins as chromatin tethers at the nuclear periphery.” Biochemical Society Transactions 39 (6): 1735–41. https://doi.org/10.1042/BST20110724.
Breslin, Thomas, Patrik Edén, and Morten Krogh. 2004. “Comparing functional annotation analyses with Catmap.” BMC Bioinformatics 5: 1–8. https://doi.org/10.1186/1471-2105-5-193.
Cai, Mengli, Y Huang, R Ghirlando, K L Wilson, R Craigie, and G M Clore. 2001. “Solution structure of the constant region of nuclear envelope protein LAP2 reveals two LEM-domain structures: one binds BAF and the other binds DNA.” The EMBO Journal 20 (16): 4399–4407. https://doi.org/10.1093/emboj/20.16.4399.
Cai, Mengli, Ying Huang, Jeong Yong Suh, John M. Louis, Rodolfo Ghirlando, Robert Craigie, and G. Marius Clore. 2007. “Solution NMR structure of the barrier-to-autointegration factor-emerin complex.” Journal of Biological Chemistry 282 (19): 14525–35. https://doi.org/10.1074/jbc.M700576200.
Clark, Neil R., Maciej Szymkiewicz, Zichen Wang, Caroline D. Monteiro, Matthew R. Jones, and Avi Ma’ayan. 2015. “Principle Angle Enrichment Analysis (PAEA): Dimensionally reduced multivariate gene set enrichment analysis tool.” In 2015 Ieee International Conference on Bioinformatics and Biomedicine (Bibm), 155:256–62. 1. IEEE. https://doi.org/10.1109/BIBM.2015.7359689.
Dinu, Irina, John D. Potter, Thomas Mueller, Qi Liu, Adeniyi J. Adewale, Gian S. Jhangri, Gunilla Einecke, Konrad S. Famulski, Philip Halloran, and Yutaka Yasui. 2007. “Improving gene set analysis of microarray data by SAM-GS.” BMC Bioinformatics 8: 1–13. https://doi.org/10.1186/1471-2105-8-242.
Edgar, R. 2002. “Gene Expression Omnibus: NCBI gene expression and hybridization array data repository.” Nucleic Acids Research 30 (1): 207–10. https://doi.org/10.1093/nar/30.1.207.
Garber, Manuel, Manfred G. Grabherr, Mitchell Guttman, and Cole Trapnell. 2011. “Computational methods for transcriptome annotation and quantification using RNA-seq.” Nature Methods 8 (6): 469–77. https://doi.org/10.1038/nmeth.1613.
Holden, Marit, Shiwei Deng, Leszek Wojnowski, and Bettina Kulle. 2008. “GSEA-SNP: applying gene set enrichment analysis to SNP data from genome-wide association studies.” Bioinformatics (Oxford, England) 24 (23): 2784–5. https://doi.org/10.1093/bioinformatics/btn516.
Irizarry, Rafael A, Chi Wang, Yun Zhou, and Terence P Speed. 2009. “Gene set enrichment analysis made simple.” Statistical Methods in Medical Research 18 (6): 565–75. https://doi.org/10.1177/0962280209351908.
Kim, Seon-Young, and David J Volsky. 2005. “PAGE: parametric analysis of gene set enrichment.” BMC Bioinformatics 6: 144. https://doi.org/10.1186/1471-2105-6-144.
Li, Junan, Anjali Mahajan, and Ming-Daw Tsai. 2006. “Ankyrin Repeat: A Unique Motif Mediating Protein−Protein Interactions †.” Biochemistry 45 (51): 15168–78. https://doi.org/10.1021/bi062188q.
Love, Michael I., Wolfgang Huber, and Simon Anders. 2014. “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biology 15 (12): 550. https://doi.org/10.1186/s13059-014-0550-8.
Maglott, D., Gota Morota, KD Pruitt, T Tatusova, P Kell, S Duvick, EP Spalding, et al. 2004. “Entrez Gene: gene-centered information at NCBI.” Nucleic Acids Research 33 (Database issue): D54–D58. https://doi.org/10.1093/nar/gki031.
Nam, Dougu, and Seon Young Kim. 2008. “Gene-set approach for expression pattern analysis.” Briefings in Bioinformatics 9 (3): 189–97. https://doi.org/10.1093/bib/bbn001.
Pan, K.-H., C.-J. Lih, and S. N. Cohen. 2005. “Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays.” Proceedings of the National Academy of Sciences 102 (25): 8961–5. https://doi.org/10.1073/pnas.0502674102.
Peng, Shaoliang, Shunyun Yang, Xiaochen Bo, and Fei Li. 2017. “paraGSEA: a scalable approach for large-scale gene expression profiling.” Nucleic Acids Research 45 (17): e155–e155. https://doi.org/10.1093/nar/gkx679.
Segura-Totten, Miriam, and Katherine L. Wilson. 2004. “BAF: roles in chromatin, nuclear structure and retrovirus integration.” Trends in Cell Biology 14 (5): 261–6. https://doi.org/10.1016/j.tcb.2004.03.004.
Simillion, Cedric, Robin Liechti, Heidi E. L. Lischer, Vassilios Ioannidis, and Rémy Bruggmann. 2017. “Avoiding the pitfalls of gene set enrichment analysis with SetRank.” BMC Bioinformatics 18 (1): 151. https://doi.org/10.1186/s12859-017-1571-6.
Snyers, Luc, Renate Erhart, Sylvia Laffer, Oliver Pusch, Klara Weipoltshammer, and Christian Schöfer. 2018. “LEM4/ANKLE-2 deficiency impairs post-mitotic re-localization of BAF, LAP2\(\alpha\) and LaminA to the nucleus, causes nuclear envelope instability in telophase and leads to hyperploidy in HeLa cells.” European Journal of Cell Biology 97 (1): 63–74. https://doi.org/10.1016/j.ejcb.2017.12.001.
Subramanian, Aravind, Heidi Kuehn, Joshua Gould, Pablo Tamayo, and Jill P. Mesirov. 2007. “GSEA-P: a desktop application for Gene Set Enrichment Analysis.” Bioinformatics 23 (23): 3251–3. https://doi.org/10.1093/bioinformatics/btm369.
Subramanian, A., P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, et al. 2005. “Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles.” Proceedings of the National Academy of Sciences 102 (43): 15545–50. https://doi.org/10.1073/pnas.0506580102.
Wagner, Nicole, and Georg Krohne. 2007. “LEM-Domain Proteins: New Insights into Lamin-Interacting Proteins.” In International Review of Cytology, 261:1–46. https://doi.org/10.1016/S0074-7696(07)61001-8.
Wang, Weichen, Zhiyi Qin, Zhixing Feng, Xi Wang, and Xuegong Zhang. 2013. “Identifying differentially spliced genes from two groups of RNA-seq samples.” Gene 518 (1): 164–70. https://doi.org/10.1016/j.gene.2012.11.045.
Wang, Xi, and Murray J. Cairns. 2014. “SeqGSEA: a Bioconductor package for gene set enrichment analysis of RNA-Seq data integrating differential expression and splicing.” Bioinformatics 30 (12): 1777–9. https://doi.org/10.1093/bioinformatics/btu090.
Yates, Bethan, Bryony Braschi, Kristian A. Gray, Ruth L. Seal, Susan Tweedie, and Elspeth A. Bruford. 2017. “Genenames.org: the HGNC and VGNC resources in 2017.” Nucleic Acids Research 45 (D1): D619–D625. https://doi.org/10.1093/nar/gkw1033.
Yoon, Sora, Seon-Young Kim, and Dougu Nam. 2016. “Improving Gene-Set Enrichment Analysis of RNA-Seq Data with Small Replicates.” Edited by Dongmei Li. PLOS ONE 11 (11): e0165919. https://doi.org/10.1371/journal.pone.0165919.
Zerbino, Daniel R., Premanand Achuthan, Wasiu Akanni, M Ridwan Amode, Daniel Barrell, Jyothish Bhai, Konstantinos Billis, et al. 2018. “Ensembl 2018.” Nucleic Acids Research 46 (D1): D754–D761. https://doi.org/10.1093/nar/gkx1098.