三、实验数据与结果
(一)rGEO——将探针对应到 HUGO 基因符号
我们实现了将 GSE 数据集中的探针统一地对应到 HUGO 基因符号的 R 包——rGEO。截至 2018 年 5 月 22 日,GSE中物种为人类的平台表格一共有 4 868种,我们获取了所有平台表格的元信息,然后用我们开发的R包进行处理,结果如图 4-A 所示。由于 GSEA 自带的基因集数据库——MSigDB——主要用于分析蛋白质编码基因,我们暂不考虑支持非编码 RNA 的基因芯片。对于五分之一左右的平台表格,rGEO 无法直接将探针对应到基因,但是可以提取出探针序列信息,在后续的改进中可以考虑通过 BLAST 到人类基因组来确定探针所对应的基因。另有六分之一左右的平台表格完全无法处理,其分为两种情况,一是根本没有任何可利用信息,一是无法通过编程来批量提取出可用信息。在数据处理过程中,我们注意到不同的平台表格在 GSE 数据集中的使用频率变异较大,于是我们以数据集为单位分析了 rGEO 的表现(每一个数据集均有一个唯一的平台表格与之对应),结果如图 4-B 所示(为了避免重复,我们舍弃了 Super Series,因为其包含的子数据集均已被纳入分析范围)。可以看出,我们开发的 R 包可以处理 GEO 中绝大部分物种为人类的数据集。
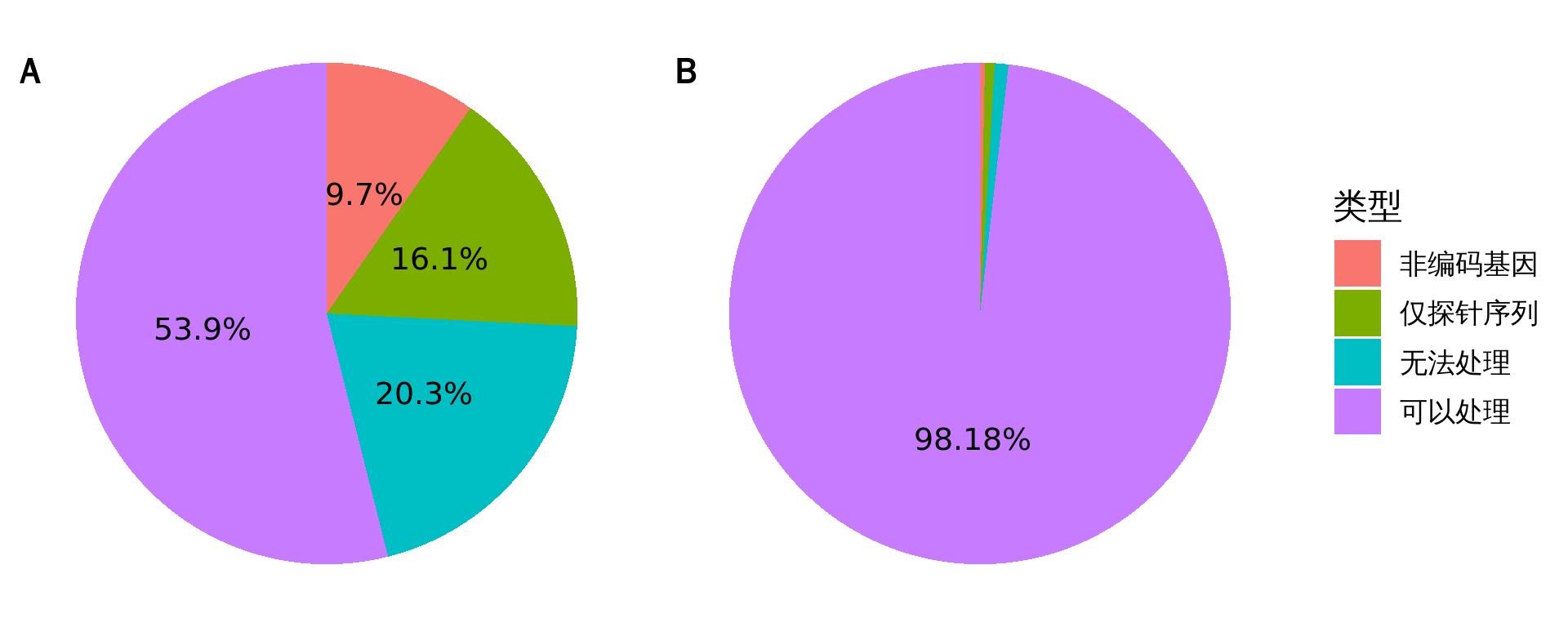
图 4: rGEO 的表现
A. 对所有物种为人类的平台表格的处理情况。B. 对所有物种为人类的数据集的处理情况。
(二)qGSEA——快捷运行 GSEA
在前文提到的 R 包的基础上,我们开发出了一个简单易用的网页应用 qGSEA,如图S1所示。其拥有用户友好的交互式界面,能够很方便地将 GSE 数据集的原始文件转换为 javaGSEA 需要的输入文件,从而使用户能快捷地使用 GSEA 这一强大的分析工具。qGSEA 有两种运行模式:A 和 B,如图 5 所示。
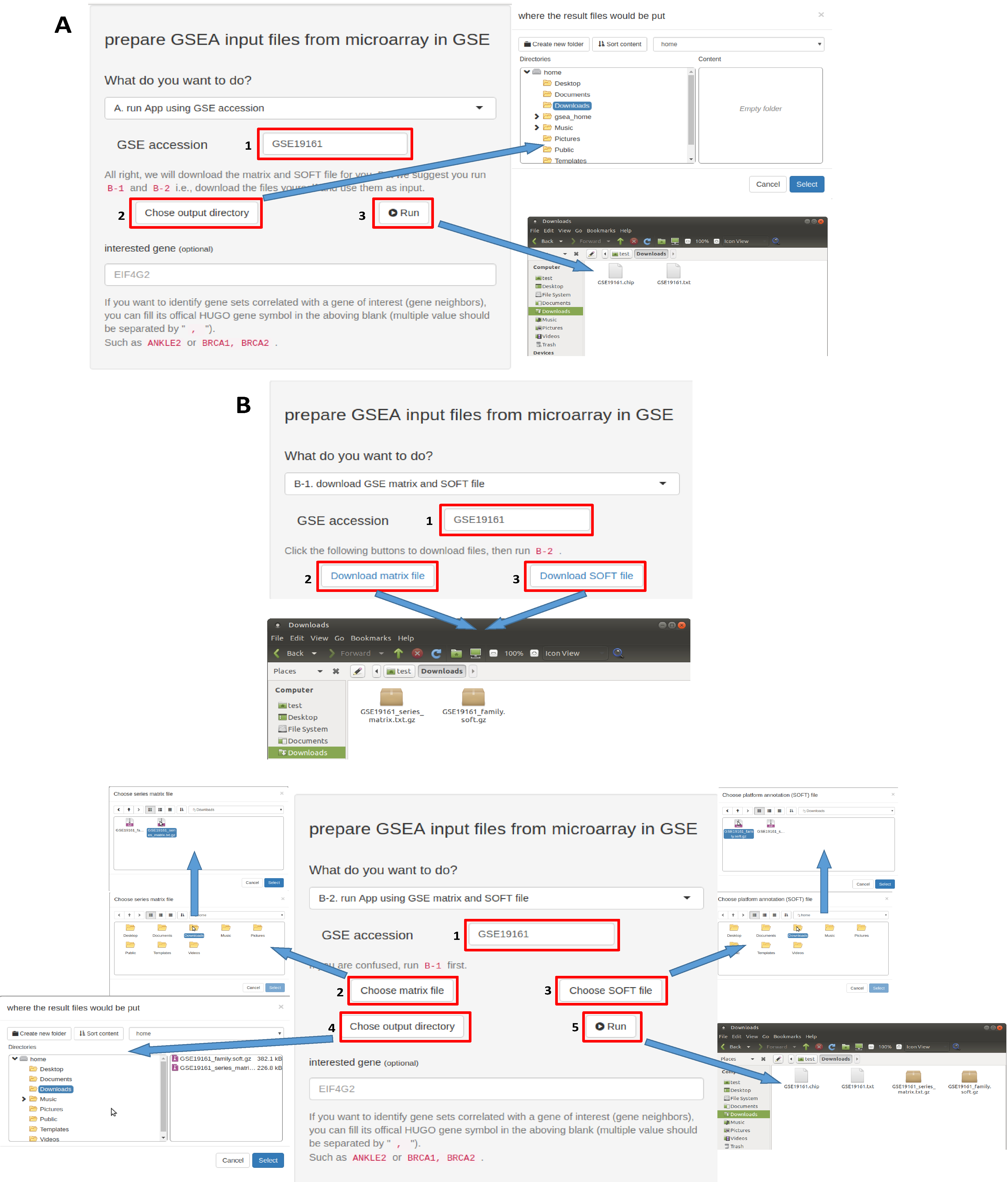
图 5: qGSEA 的使用方法
A. 模式 A 的操作步骤。B. 模式 B-1 的操作步骤。C. 模式 B-2 的操作步骤。
对于模式 A,用户需要输入 GSE accession,然后选择存放输出文件的文件夹,最后点击 Run,即可在之前选择的文件夹中得到符合 javaGSEA 规范的输入文件,如图 5-A 所示。在实际使用过程中,我们发现从 NCBI 下载数据十分缓慢,当文件较大时需要耗费较长时间,而且网络连接非常不稳定,有时甚至会完全卡死。基于这种情况,我们提供了另一种运行方法——模式 B——先由用户下载 GSE 的原始数据文件,然后再由这些文件得到 javaGSEA 需要的输入文件。
模式 B 分为 B-1、B-2 两步。在 B-1 中,用户需要输入 GSE accession,然后单击下方的两个按钮即可下载 GSE 的原始数据文件——matrix 文件和 SOFT 文件,如图 5-B所示。此时文件是通过浏览器来下载,用户可以查看下载进度,在下载失败后重新下载,或者复制下载链接之后通过其它方式来下载(比如使用专门的下载软件,或者等到网络环境较好的时候再下载)。不管通过何种方式,在用户得到 GSE 的原始数据文件之后,即可运行 B-2。用户仍需输入 GSE accession(如果在 B-1中已经输入过,则此时会被保留,无需重复输入),然后分别选中下载好的 matrix 文件和 SOFT 文件,以及输出文件夹,最后点击 Run 即可得到与模式A一样的结果,如图 5-C所示。
除了上述提到的必要步骤之外,在 A 和 B-2 中还提供了一项可选步骤。当用户希望使用 GSEA 筛选出与某一感兴趣的基因的表达相关联的基因集时,可以在最下方的框中输入该基因的 HUGO 基因符号。这样 qGSEA 就会额外生成一个 .cls 文件,在 javaGSEA 中该文件用于提供表型标签。
运行完qGSEA之后的任务就十分简单明了了,用户只需要把生成的文件拖拽到 javaGSEA 中,然后按照其指示即可得到分析结果。
qGSEA 的一大亮点是,在软件运行的整个过程中都会对用户的错误给出简短、清晰的提示,当用户修正该错误后提示会立即消失,在用户犯了新的错误后又会给出新的提示,从而引导用户一步一步达成运行软件的目的,如图 S2 所示。这些提示进一步增强了 qGSEA 的易用性,即使是第一次接触 qGSEA 的用户,也能够在指示的引导下正确地运行软件。而且在用户修正所有的错误之前,qGSEA 会拒绝运行,这样就大大减少了产生错误输出的可能性。
(三)在883项数据集中运行GSEA并整合结果
1. 运行GSEA
我们从 GEO 数据库获取了 1 187项人类乳腺癌或卵巢癌相关的数据集,然后使用 qGSEA 从原始数据文件生成 javaGSEA 输入文件。其中有 21 项无法被 qGSEA 识别,剩下的 1 166项数据集中只有 883 项包含了 LEM4 基因,我们在这些数据中进行了 GSEA。
由于我们使用重排表型(1 000次)的方式计算 P 值,为了实现足够的分辨率,每一数据集样本数至少为 7。我们发现样本数多的数据集倾向于产生更多统计显著的结果(数据未显示),这可能是因为大样本的数据集有着更高的信噪比。考虑到以上两点,我们仅选取样本数超过某一阈值的数据集的结果进行后续分析,但是阈值设置过高又会导致可用的数据集太少,以至于损失过多有用信息。权衡利弊之后,我们选取样本数超过 100(包含 100)的数据集(共 108 项)。
2. 整体分析GSEA的结果
我们分析了不同集合整体的情况,如图 6 所示。我们认为 c3.mir、c4.cgn 和 c7这3个集合表现比较奇怪。在正常情况下,每个数据集中应该只有一小部分统计显著的基因集,在其它集合中,绝大部分数据集都与该情况相符。但是在 c3.mir、c4.cgn 和 c7 中,有相当一部分数据集都给出了很多(甚至绝大部分都是)统计显著的基因集。我们认为造成该现象的原因有两种,一是该集合中的基因集组成不合理,容易产生假阳性;一是 LEM4 确实能通过某种生物学机制影响到该集合中的大部分基因集。针对 c3.mir,LEM4 部分定位于内质网上,在维持内质网的形态中起到重要的作用,而内质网又是 microRNA 加工的重要场所,我们推测 LEM4 可能会通过影响内质网的稳定性来影响 microRNA 的加工,从而影响 c3.mir 中的大部分基因集。针对 c4.cgn,实验室前期研究发现 LEM4 在乳腺癌和卵巢癌中是重要的癌基因,能够影响抑癌基因 RB1 和原癌基因 MYC,我们推测 LEM4 可能通过癌基因之间的关系网络来影响 c4.cgn 中的大部分基因集。至于 LEM4 与 c7 集合的关系,目前还不清楚。
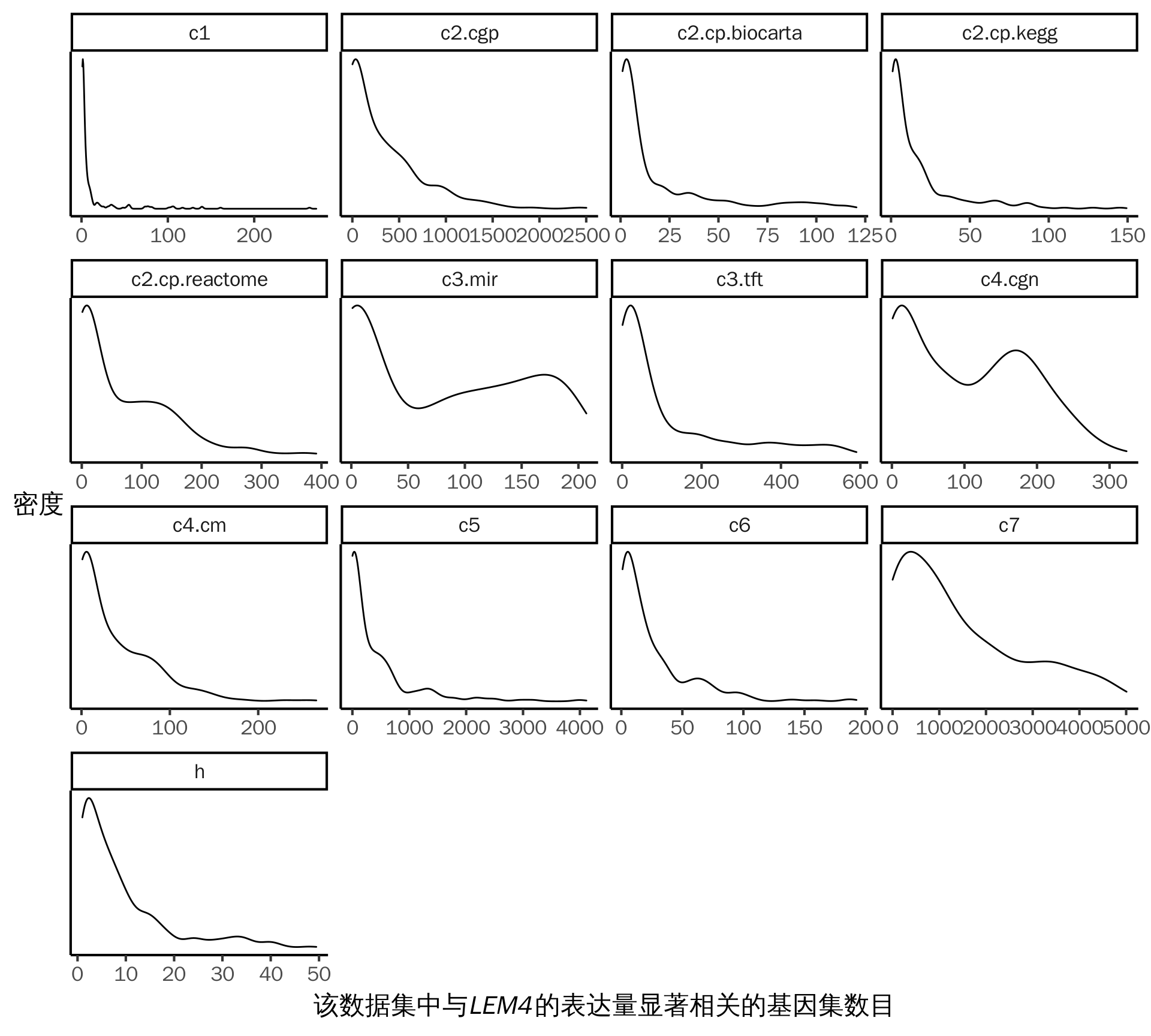
图 6: 各个集合在不同数据集中的整体表现
接下来,我们希望筛选出与 LEM4 相关性最强的基因集。我们将不同数据集的结果整合在一起,发现它们之间的一致性较差,即统计显著的基因集重合度较低。于是我们使用某一基因集表现为统计显著的数据集数目n,来衡量该基因集与 LEM4 的相关程度(具体计算方法见“材料与方法”部分)。我们注意到并非每一个基因集都在所有数据集中进行了 GSEA,于是我们计算出了某一基因集进行了 GSEA 的数据集数目 N。这一现象意味着在评估 n 时,N 也是一个不可忽略的因素,因为如果一个与 LEM4 真正相关的基因集只在较少数据集中进行了 GSEA,那么与在较多数据集中进行了 GSEA 的噪音基因集相比,前者的 n 值可能反而会更低。
为了找出影响 N 的因素,我们检查了 GSEA 的输出日志,发现上述基因集在特定数据集中被包含的基因太少,以至于无法进行富集分析。由于基因芯片在本质上就不一定能完全覆盖转录组,而且在基因注释等处理环节可能会产生疏漏,我们决定不筛除对转录组覆盖率低的数据集,以避免损失有效信息。一种可行的改进方法是,用 n 与 N 的比值作为衡量基因集与 LEM4 相关性的指标。但是这样做会带来另一个问题,即当一个基因集的 N 较小时,难以区分随机因素与生物学效应对该指标的影响。考虑到我们的主要目的是发现新的研究靶点,我们决定牺牲灵敏度、确保特异度,仍然使用 n 作为主要评估指标,同时给出 N 作为决策的参考依据。为避免数据异质性的影响,我们排除了 c3.mir、c4.cgn 和 c7 这三个集合,同时排除了其它集合中包含过多(25% 及以上)统计显著的基因集的数据集。
由于每个集合中都有很多 n 值很小的基因集,为了更好地展示数据分布,我们从每个集合中分别选取前 10 位正相关和前 10 位负相关的基因集,如图 7 所示。可以看出不同集合之间存在明显差异,而且正相关的基因集的 n 值明显大于负相关。为了探索其原因,我们推测该现象可能与 LEM4 基因本身的性质有关,于是我们分析了 BRCA1、BRCA2、TP53、MYC 和 NOTCH1 这5个基因(数据未显示),均发现了类似的趋势,暗示 GSEA 这一分析本身可能存在偏差。接下来的分析主要专注于正相关的基因集。
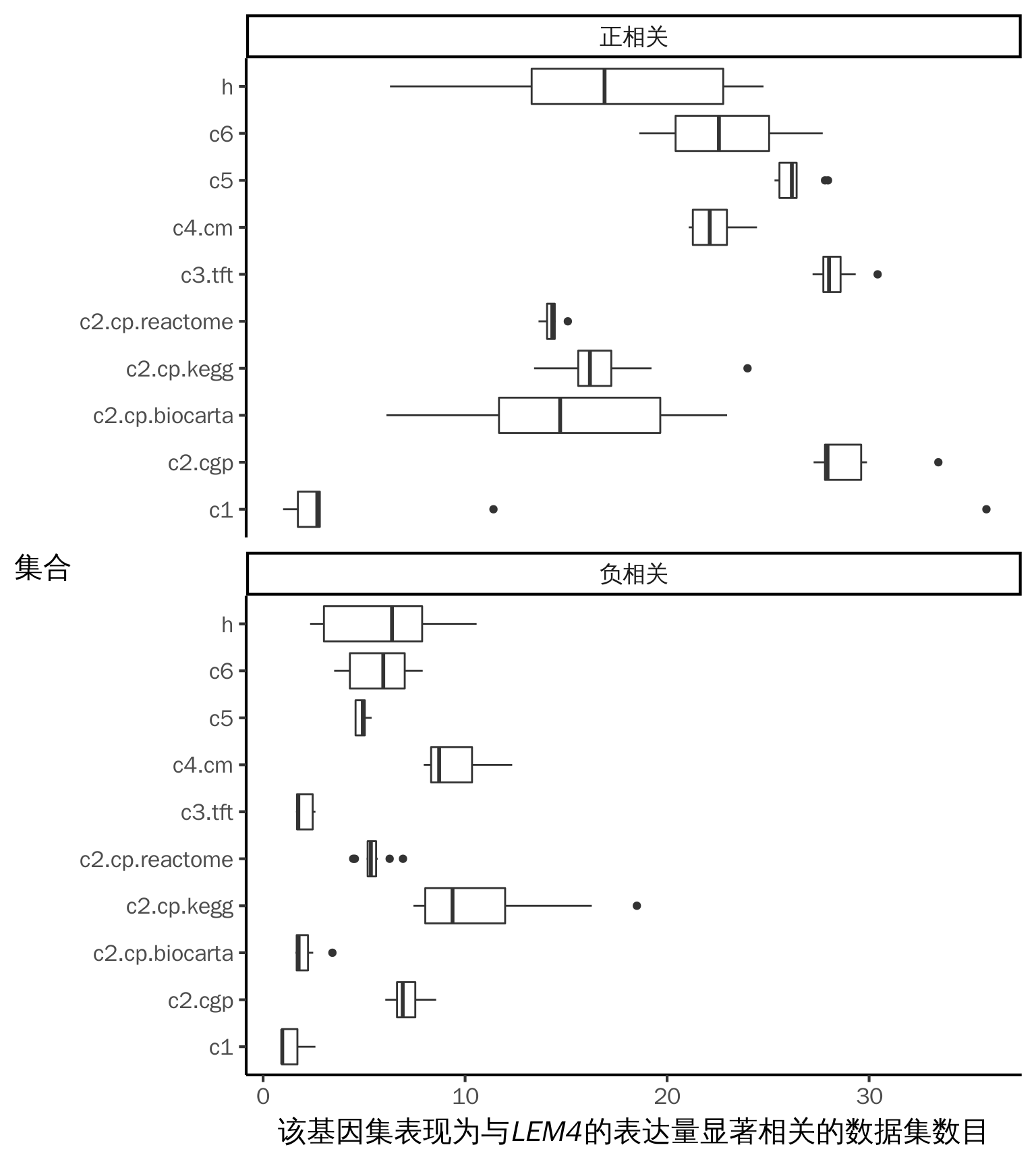
图 7: 各个集合中前 10 位正相关和前 10 位负相关的基因集
3. 探索筛选基因集的方法
为了筛选出与 LEM4 相关性最强的基因集,一个直观的想法也许是取 n 排名靠前(如前 25)的基因集,如表 S1 所示。但是这样会导致有些集合会有较多基因集入选,而另一些则较少,例如有 10 个基因集来自 c3.tft,而来自 h 的基因集一个都没有。为了应对这一问题,一种方案是同时保留整体排名靠前(如前 15)和在各自集合中分别排名靠前(如前 2)的基因集。为了评价该方案的合理性,我们对 c1、c3.tft 和 h 这三个集合进行了深入探索,如表 S2 所示。
c1 集合整体表现不是很好,但是其中最靠前的基因集 CHR12Q24(n=36)恰好与 c2.cgp 中排名第 5 的基因集 NIKOLSKY_BREAST_CANCER_12Q24_AMPLICON(n=28)相符合(见表 S1),后者是 Nikolsky et al. (2008) 在乳腺癌中鉴定出一个扩增子(amplicon)。而排名第二的基因集 CHR12Q23 的 n 值则仅为 11(见表S2),可能仅仅只是因为与 CHR12Q24 距离较近。后面的基因集的 n 值就都不怎么突出了。
c3.tft 这一集合中,排名前 10 的基因集均为 E2F 转录因子家族。这一结果确实得到了实验室前期实验证据的直接支持,但是冗余度太高。
h 集合中排名前 8 位的基因集的 n 值均较为突出,但是我们发现前 5 位和第 6 至 8 位之间存在明显的分界线。从生物学意义来看,E2F_TARGETS、MYC_TARGETS_V1 和 MYC_TARGETS_V2 均能被实验室前期的实验证据直接支持,而 MITOTIC_SPINDLE 和 G2M_CHECKPOINT 则与 LEM4 在细胞周期中重要作用吻合,但是 MTORC1_SIGNALING、DNA_REPAIR 和 UNFOLDED_PROTEIN_RESPONSE 则与 LEM4 目前已知的功能几乎没有关系(基因集名称均省略了前缀 HALLMARK_)。结合以上两点,我们认为第 6 至 8 位基因集是假阳性结果,其原因可能是与前 5 位的基因集有较多重合的基因。为了验证这一点,我们利用 MSigDB 提供的重合分析工具(http://software.broadinstitute.org/gsea/msigdb/help_annotations.jsp#overlap)搜索了这三个基因集,如图 8 所示,可以看出它们分别与前 5 的基因集中的 2、2 和 4 个有明显的重合。作为对照,对于每个基因集,我们都从 h 集合中选取了相同类别且大小相近的两个基因集进行同样的搜索,如图 S3 所示,共计 6 个基因集中只有 1 个与前 5 的基因集中的 1 个有明显重合(另有 1 个与前 5 的基因集中的 1 个有不太明显的重合),从而验证了我们的想法。
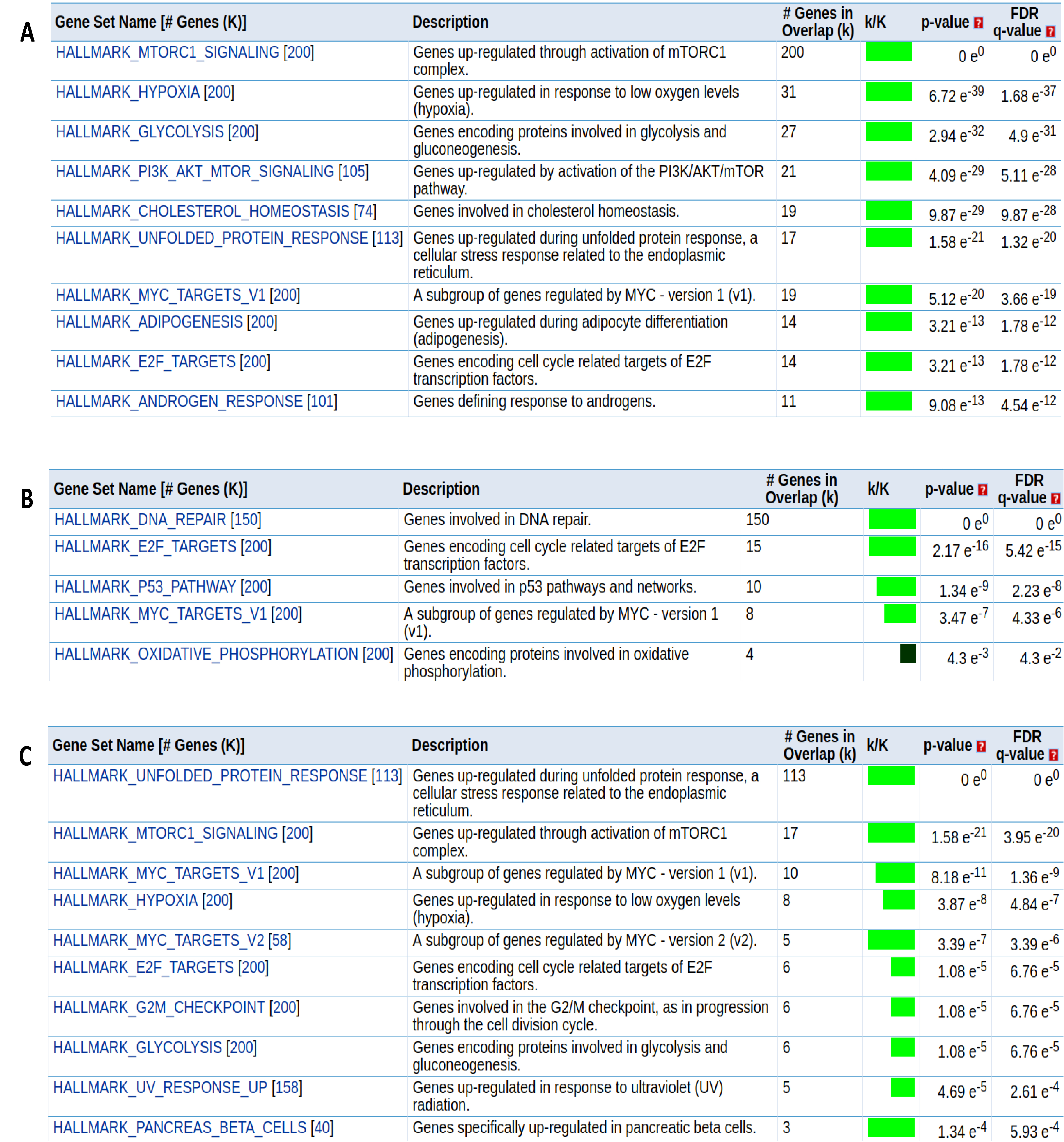
图 8: h 集合中第 6 至 8 位基因集均与某些前 5 位基因集存在明显重合
A. HALLMARK_MTORC1_SIGNALING。B. HALLMARK_DNA_REPAIR。 C. HALLMARK_UNFOLDED_PROTEIN_RESPONSE。
4. 筛选最终结果并分析其生物学意义
根据前文的探究,我们觉得应该针对每个集合的情况进行具体分析,既要选出该集合中 n 值最突出的基因集(与其它基因集存在明显的差异),也要考虑该集合能提供的生物学信息。如果对所有集合都采用同一标准(比如取前 2 位基因集)的话,在某些集合(比如 h)中会损失重要的信息,在某些集合(比如 c3.tft)中又产生会多余的信息。
具体来说,我们从每个集合中筛选出 n 值最突出的少量基因集(一般不超过 3 个,考虑到 h 集合的特殊性,我们选取了 5 个),作为核心基因集,用于产生生物学假设,如表 S3 所示。从其它排名靠前的基因集中挑出与核心基因集有关的部分,作为补充基因集,用于支持和具化假设。
核心基因集中有一些能被实验室前期的研究或已发表的论文直接支持,包括参与有丝分裂后核膜重建、调控细胞周期蛋白 D-CDK4/6-Rb 轴和调控 ERα 信号通路,如表 2 所示(能够支持这些功能的补充基因集如表 S4 所示)。这说明了我们的分析流程的合理性,也暗示核心基因集中的其它成员很可能揭示了 LEM4 尚未被发现的其它功能。
| 集合 | 基因集 | N | n |
|---|---|---|---|
| LEM4 已知的生物学功能 | |||
| (1)参与有丝分裂后核膜重建 | |||
| c5 | GO_NUCLEAR_ENVELOPE_ORGANIZATION |
86 | 28 |
| (2)调控细胞周期蛋白 D-CDK4/6-Rb 轴 | |||
| h | HALLMARK_E2F_TARGETS |
71 | 23 |
| c2.cp.biocarta | BIOCARTA_G1_PATHWAY |
79 | 23 |
| c3.tft | E2F_Q3_01 |
72 | 30 |
| c6 | RB_P107_DN.V1_UP |
76 | 28 |
| (3)调控 ERα 信号通路 | |||
| h | HALLMARK_MYC_TARGETS_V1 |
67 | 22 |
| h | HALLMARK_MYC_TARGETS_V2 |
73 | 19 |
| LEM4 可能的新的生物学功能 | |||
| (1)在 G2 期到 M 期的转化中起到关键作用 | |||
| h | HALLMARK_G2M_CHECKPOINT |
76 | 24 |
| c2.cp.biocarta | BIOCARTA_G2_PATHWAY |
79 | 22 |
| (2)通过影响微管蛋白,从而影响纺锤体的形成 | |||
| h | HALLMARK_MITOTIC_SPINDLE |
71 | 25 |
| (3)调控 S 期 CDK,从而促进 DNA 复制 | |||
| c2.cp.biocarta | BIOCARTA_MCM_PATHWAY |
79 | 20 |
| (4)直接影响核孔的 RNA 转运功能 | |||
| c2.cp.reactome | REACTOME_TRANSPORT_OF_MATURE_TRANSCRIPT_TO_CYTOPLASM |
72 | 15 |
| c5 | GO_NUCLEAR_PORE |
84 | 28 |
N,某一基因集进行了GSEA的数据集数目,n,某一基因集表现为统计显著的数据集数目(后文的表格中不再解释)。
在此基础上,我们从其它核心基因集中筛选出与 LEM4 已知的生物学功能相符合的部分,从中总结出 4 项 LEM4 可能的新功能,如表 2 所示。其中一些功能得到了补充基因集的支持,另一些则依赖补充基因集来启发我们具体的作用机制,如表 3 所示。关于 LEM4 影响纺锤体的方式,我们认为其影响了微管的聚合和解聚;关于 LEM4 对核孔的影响,我们根据多个与RNA转运有关的基因集,猜想 LEM4 除了影响核膜形成之外,会能直接与核孔复合体相互作用,影响 RNA(包括 mRNA 和 tRNA)的转运。
| 集合 | 基因集 | N | n |
|---|---|---|---|
| (1)在 G2 期到 M 期的转化中起到关键作用 | |||
| c2.cp.reactome | REACTOME_MITOTIC_G2_G2_M_PHASES |
66 | 14 |
| (2)通过影响微管蛋白,从而影响纺锤体的形成 | |||
| c5.all | GO_REGULATION_OF_MICROTUBULE_POLYMERIZATION_OR_DEPOLYMERIZATION |
85 | 26 |
| c5.all | GO_REGULATION_OF_MICROTUBULE_BASED_PROCESS |
85 | 25 |
| c5.all | GO_MITOTIC_SPINDLE |
82 | 25 |
| (3)调控 S 期CDK,从而促进 DNA 复制 | |||
| c5.all | GO_ATP_DEPENDENT_DNA_HELICASE_ACTIVITY |
89 | 26 |
| c5.all | GO_DNA_HELICASE_ACTIVITY |
91 | 26 |
| (4)直接影响核孔的 RNA 转运功能 | |||
| c2.cp.reactome | REACTOME_TRANSPORT_OF_MATURE_MRNA_DERIVED_FROM_AN_INTRONLESS_TRANSCRIPT |
64 | 14 |
| c5.all | GO_TRNA_TRANSPORT |
83 | 25 |
References
Nikolsky, Yuri, Evgeny Sviridov, Jun Yao, Damir Dosymbekov, Vadim Ustyansky, Valery Kaznacheev, Zoltan Dezso, et al. 2008. “Genome-wide functional synergy between amplified and mutated genes in human breast cancer.” Cancer Research 68 (22): 9532–40. https://doi.org/10.1158/0008-5472.CAN-08-3082.